21/01/2024
A jornada de um medicamento dentro do corpo humano é um processo complexo e fascinante, fundamental para sua eficácia terapêutica. Compreender como os fármacos são distribuídos e, em particular, como interagem com o sistema nervoso central (SNC), é crucial tanto para profissionais de saúde quanto para o público em geral. Este artigo explora em profundidade o conceito de distribuição de medicamentos, detalhando sua trajetória desde a corrente sanguínea até os tecidos, e mergulha no universo dos medicamentos que atuam no SNC, abordando suas classificações, mecanismos de ação e implicações clínicas.
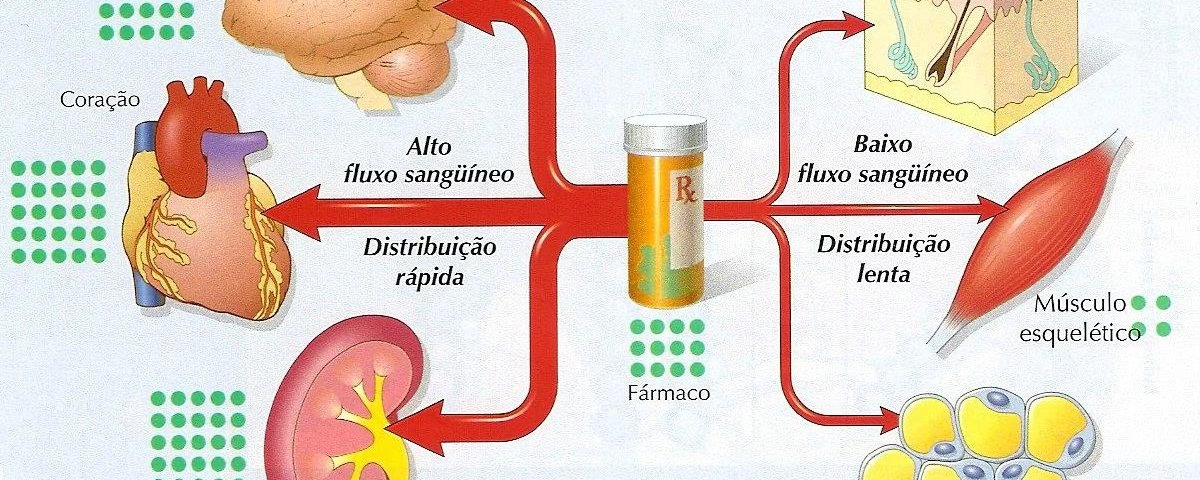
A distribuição de medicamentos é o movimento intrínseco de uma substância do sangue para os diversos tecidos do corpo — como o tecido adiposo, muscular e cerebral — e, inversamente, dos tecidos de volta para o sangue. Este processo também envolve a determinação das proporções relativas do medicamento em cada tecido. É uma fase crítica após a absorção do fármaco na corrente sanguínea. Uma vez absorvido, o medicamento circula rapidamente pelo corpo; em média, o sangue completa uma circulação em aproximadamente um minuto. Graças a essa recirculação constante, o medicamento é transportado eficientemente da corrente sanguínea para os tecidos.
É importante notar que a distribuição da maioria dos medicamentos não ocorre de maneira uniforme por todo o corpo. Fármacos que se dissolvem facilmente em água, conhecidos como medicamentos hidrossolúveis (como o atenolol, um anti-hipertensivo), tendem a permanecer predominantemente no sangue e no líquido que circunda as células, denominado espaço intersticial. Em contrapartida, os medicamentos solúveis em gordura, ou lipossolúveis (a exemplo do clorazepato, um ansiolítico), demonstram uma maior tendência a se concentrar nos tecidos adiposos. Além disso, alguns medicamentos possuem uma afinidade especial por certas regiões do corpo, concentrando-se principalmente em uma pequena parte, como o iodo que se acumula na glândula tireoide, devido à capacidade desses tecidos de reter o fármaco.
A velocidade com que os medicamentos penetram em diferentes tecidos varia significativamente, dependendo de sua capacidade de atravessar as membranas celulares. Por exemplo, a rifampicina, um antibiótico altamente lipossolúvel, consegue penetrar o cérebro rapidamente, enquanto a penicilina, um antibiótico hidrossolúvel, não o faz com a mesma facilidade. Em geral, medicamentos lipossolúveis atravessam as membranas celulares de forma mais eficiente do que os hidrossolúveis. Em alguns casos, mecanismos de transporte específicos auxiliam o movimento de entrada e saída dos fármacos dos tecidos, garantindo que cheguem aos seus alvos ou sejam eliminados.
A ligação de medicamentos às proteínas circulantes no sangue é outro fator determinante na sua distribuição. Alguns fármacos se ligam fortemente a essas proteínas, o que faz com que saiam da corrente sanguínea muito lentamente. Outros, por sua vez, ligam-se mais fracamente, permitindo que saiam rapidamente e penetrem em outros tecidos. É comum que uma parte, ou até mesmo a maioria, das moléculas de um medicamento no sangue se unam às proteínas sanguíneas. A porção ligada à proteína é geralmente inativa. À medida que a parte não ligada se distribui pelos tecidos e seu nível na corrente sanguínea diminui, as proteínas sanguíneas liberam gradualmente o medicamento ligado a elas. Dessa forma, o medicamento ligado atua como um verdadeiro reservatório de fármaco na corrente sanguínea, garantindo um efeito prolongado.
Certos medicamentos também podem se acumular em tecidos específicos, servindo como reservatórios de excesso de fármaco. Por exemplo, a digoxina, um medicamento para o coração, acumula-se no coração e nos músculos esqueléticos. Esses tecidos liberam lentamente o medicamento de volta para a corrente sanguínea, o que evita uma redução abrupta dos níveis sanguíneos e, consequentemente, prolonga o efeito terapêutico do medicamento. Em casos de fármacos que se acumulam nos tecidos adiposos, a liberação pode ser tão lenta que eles continuam a circular na corrente sanguínea por dias após a interrupção do tratamento.
É importante ressaltar que a distribuição de um medicamento pode variar consideravelmente de pessoa para pessoa. Indivíduos com obesidade, por exemplo, podem armazenar uma quantidade significativamente maior de medicamentos lipossolúveis em seus tecidos adiposos, enquanto pessoas muito magras armazenam uma quantidade menor. Curiosamente, adultos mais velhos, mesmo que não sejam obesos, podem acumular uma grande quantidade de medicamentos lipossolúveis devido ao aumento da proporção de gordura corporal que ocorre naturalmente com o envelhecimento.
- Fármacos com Ação no Sistema Nervoso Central (SNC)
- Neurotransmissão no Sistema Nervoso Central
- Fármacos Antidepressivos
- Fármacos Antipsicóticos
- Fármacos Ansiolíticos, Sedativos e Hipnóticos
- Tratamentos dos Distúrbios Degenerativos do SNC
- Fármacos Anticonvulsivantes
- Anestésicos Gerais
- Tabela Comparativa de Antidepressivos
- Perguntas Frequentes sobre Medicamentos do SNC
Fármacos com Ação no Sistema Nervoso Central (SNC)
Os fármacos que atuam no sistema nervoso central possuem uma relevância imensa no tratamento de uma vasta gama de distúrbios psiquiátricos, como depressão, ansiedade e esquizofrenia, além de condições neurológicas degenerativas e epilepsia. No Brasil, a comercialização desses fármacos é estritamente regulada pela Portaria nº 344/1998 da Agência Nacional de Vigilância Sanitária (ANVISA), que os classifica como psicotrópicos e entorpecentes. Compreender como esses fármacos atuam, quais sistemas de neurotransmissão estão envolvidos e seus principais aspectos farmacológicos é fundamental para um uso seguro e eficaz.
Neurotransmissão no Sistema Nervoso Central
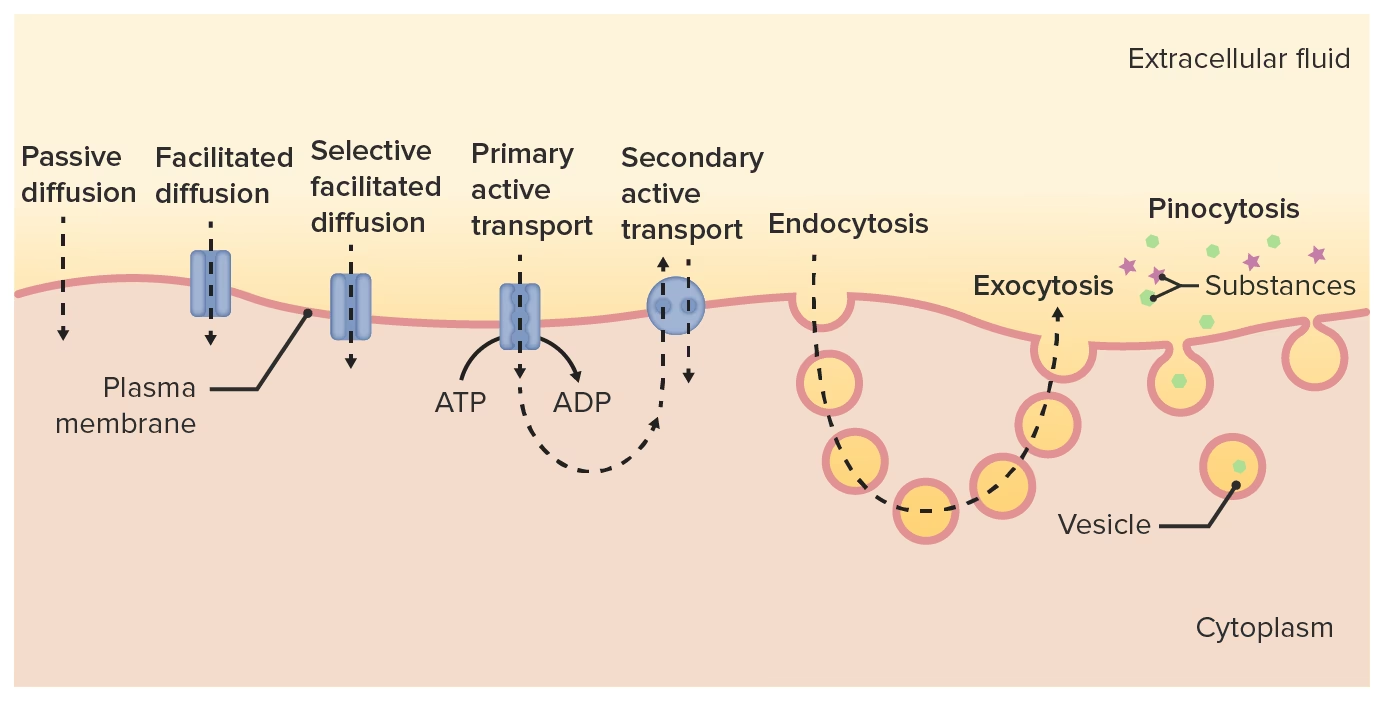
A base da ação dos fármacos no SNC reside na alteração da neurotransmissão. Neurotransmissores são pequenas moléculas sinalizadoras, sintetizadas nos neurônios, que transmitem informações fisiológicas a outros neurônios, glândulas ou músculos. Eles atuam estimulando ou inibindo uma transmissão nervosa após a ativação de um receptor específico no neurônio pós-sináptico. Os neurônios pré-sinápticos sintetizam e armazenam os neurotransmissores em vesículas nas terminações sinápticas. Após um estímulo, um potencial de ação é deflagrado, levando à exocitose (liberação) dos neurotransmissores na fenda sináptica.
No neurônio pós-sináptico, os neurotransmissores ativam canais iônicos regulados por ligante ou receptores acoplados à proteína G, desencadeando uma resposta. O efeito dessas moléculas é tipicamente abreviado por mecanismos como a recaptação por uma proteína transportadora presente na membrana do neurônio pré-sináptico, a inativação enzimática na própria fenda sináptica, ou a captação e degradação por uma célula glial adjacente. É crucial entender que o efeito de qualquer neurotransmissor é devido à sua liberação local, pois eles não conseguem atravessar a barreira hematoencefálica.
Alguns dos principais neurotransmissores do SNC incluem:
- Acetilcolina: Essencial para a manutenção do alerta e vigília, facilita a excitabilidade do córtex cerebral e modula o processamento sensorial, além de participar ativamente dos mecanismos de memória e aprendizagem.
- Noradrenalina: Influencia o sistema de alerta e vigília, mantém os processos de atenção, participa de respostas emocionais (aversivas como raiva e agressão, e gratificantes como afeto) e no controle da fome e saciedade.
- Glutamato: O principal neurotransmissor excitatório do SNC, envolvido desde a ativação mínima necessária para manter uma via de transmissão aberta até a excitação persistente que pode culminar em processos patológicos.
- Dopamina: Fundamental para o controle motor voluntário, participa em processos onde a motivação é essencial para o comportamento, e contribui para manter a atenção, a avaliação correta da realidade e o controle do pensamento.
Os principais alvos dos fármacos psicotrópicos no SNC são: (1) receptores específicos de neurotransmissores, (2) transportadores de membrana e (3) enzimas, cuja inibição pode aumentar a concentração de neurotransmissores na fenda sináptica.
Fármacos Antidepressivos
A depressão maior, ou depressão unipolar, é uma condição clínica incapacitante, relativamente comum, de caráter crônico e recorrente em aproximadamente 80% dos casos. Embora mais prevalente em mulheres, continua sendo uma doença subdiagnosticada e subtratada. Outras apresentações com sintomas menos intensos, como a distimia, são igualmente frequentes. A utilização de antidepressivos pode melhorar ou eliminar os sintomas da depressão moderada a grave. Contudo, antes de iniciar o tratamento, é imperativo descartar o diagnóstico de transtorno bipolar, já que antidepressivos podem desencadear o aparecimento de episódios de mania.
As principais classes de fármacos antidepressivos são os inibidores da monoamino-oxidase (IMAO), os antidepressivos tricíclicos (ADT) e os inibidores seletivos da recaptação de serotonina (ISRS).
Inibidores da Monoamino-Oxidase (IMAO)
A enzima monoamino-oxidase (MAO) está localizada na membrana mitocondrial externa e é expressa na maioria dos tecidos corporais, com altos níveis no fígado. Existem duas isoformas da MAO: a MAO-A, que catalisa a degradação da serotonina (5-HT) e noradrenalina (NA), e a MAO-B, com maior seletividade por benzilaminas e 2-feniletilaminas. Fármacos que inibem a MAO-A mostram-se eficazes como antidepressivos, enquanto os inibidores seletivos da MAO-B são utilizados principalmente para tratar a doença de Parkinson.
O efeito farmacológico dos IMAO está relacionado com o aumento da capacidade de armazenamento e liberação dos neurotransmissores 5-HT e NA pelos neurônios no SNC. Após a liberação na fenda sináptica, esses neurotransmissores são recaptados por transportadores de membrana. No neurônio pré-sináptico, a MAO atua controlando a quantidade de monoaminas no terminal axônico. Na presença dos IMAO, observa-se um aumento da concentração citoplasmática de 5-HT e NA nas terminações nervosas e da taxa de liberação espontânea desses neurotransmissores.
O principal efeito adverso observado com os IMAO é a hipotensão, decorrente da diminuição do tônus simpático e consequente redução da pressão arterial sistêmica. Outros efeitos adversos comuns incluem tremores, insônia, ganho de peso (devido ao aumento do apetite) e efeitos anticolinérgicos, como boca seca, visão embaçada e retenção urinária. A síndrome serotonínica, uma condição grave, pode ocorrer devido a um excesso de 5-HT na fenda sináptica, provocando confusão, excitação, hiperatividade neuromuscular, hiper-reatividade autonômica e hipertermia.
Antidepressivos Tricíclicos (ADT)
Os ADT têm uma ampla gama de usos, além do tratamento da depressão, incluindo dor neuropática, enxaqueca, transtorno obsessivo compulsivo (TOC), transtorno do déficit de atenção com hiperatividade (TDAH), vômitos e insônia. O mecanismo pelo qual os ADT melhoram o quadro depressivo está relacionado ao aumento da concentração de 5-HT e NA nas fendas sinápticas do SNC, pois inibem a recaptação desses neurotransmissores pelos neurônios pré-sinápticos.
Além disso, a maioria dos ADT atua como antagonistas de diversos receptores pós-sinápticos, como H1, 5-HT2A, α1-adrenérgico e muscarínicos, cujas ações podem ser benéficas ou indesejáveis. Todos os ADT, sendo a doxepina o mais potente, atuam como antagonistas H1, produzindo sedação e adicionando um efeito terapêutico. O antagonismo do receptor 5-HT2A (por doxepina, amitriptilina e clomipramina) pode ser clinicamente relevante na melhora do sono. O antagonismo do receptor α1-adrenérgico, similar em todos os ADT, é responsável pela indução da hipotensão ortostática ou postural, o principal efeito indesejável desses fármacos. Os efeitos anticolinérgicos, como boca seca, visão borrada, retenção urinária, constipação e prejuízo sobre a memória (que pode resultar em delírio), estão relacionados ao antagonismo muscarínico.
A interação medicamentosa mais grave dos ADT é o uso associado ao etanol, que pode induzir uma depressão respiratória grave com consumo excessivo de bebidas alcoólicas. Os ADT também interferem na ação de muitos fármacos anti-hipertensivos. Devido à potencialização das aminas simpaticomiméticas pelos ADT, deve-se evitar o uso concomitante com fármacos IMAO.
Inibidores Seletivos da Recaptação de Serotonina (ISRS)
Os ISRS são, na maioria dos casos, a primeira escolha no tratamento da depressão maior, principalmente devido à sua melhor margem de segurança em comparação com os ADT e IMAO. Além da depressão, os ISRS são indicados para o tratamento da ansiedade generalizada, síndrome do pânico, transtorno obsessivo compulsivo e distúrbios alimentares. Como esses fármacos não bloqueiam os receptores H1, α1-adrenérgicos e muscarínicos, os efeitos indesejados são menos intensos, o que favorece a adesão do paciente ao tratamento.
Os efeitos colaterais mais comuns dos ISRS incluem distúrbios gastrintestinais, ansiedade, disfunção sexual, prejuízo da cognição, possibilidade de desenvolver síndrome serotonínica e ideação suicida. Apesar desses potenciais efeitos, a segurança relativa dos ISRS os torna uma opção amplamente preferida na prática clínica moderna.
Fármacos Antipsicóticos
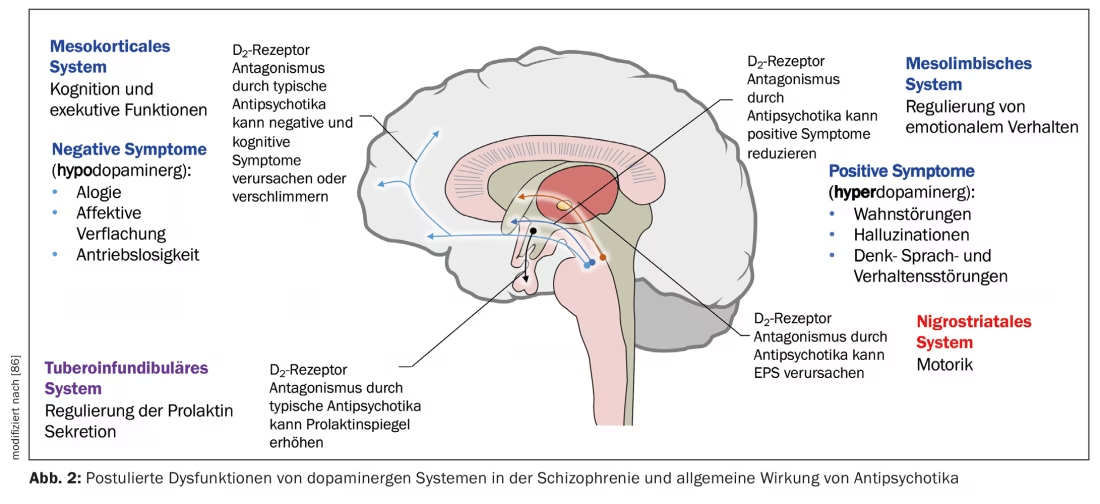
Os transtornos psicóticos mais comuns incluem mania (transtorno bipolar), psicose induzida por substâncias (por exemplo, cocaína) e esquizofrenia. Os fármacos disponíveis para tratar essas condições são classificados como antipsicóticos típicos, atípicos e estabilizadores do humor, sendo os últimos indicados principalmente no tratamento da mania ou transtorno bipolar.
Antipsicóticos Típicos
Os antipsicóticos típicos induzem seu efeito farmacológico pelo antagonismo dos receptores D2 pós-sinápticos no prosencéfalo. O tratamento crônico com esses fármacos requer cuidado devido à extensão dos efeitos colaterais observados. A principal característica dos antipsicóticos típicos é a capacidade de induzir sintomas extrapiramidais, relacionados ao bloqueio dopaminérgico, sendo mais proeminente com o haloperidol. Os principais efeitos adversos neurológicos agudos incluem acatisia (incapacidade de se manter parado), parkinsonismo e distonia, que geralmente diminuem ou desaparecem com a redução da dose.
Antipsicóticos Atípicos
Os antipsicóticos atípicos, ou de segunda geração, promovem um antagonismo D2 com menor potência, mas têm a capacidade de interagir com outros receptores, variando muito entre os fármacos dessa classe. Por exemplo, a clozapina, além de interagir com D2, antagoniza também outros receptores para DA (D1, D3 e D4), histamina (H1), muscarínicos (M1), serotonina (5-HT2A, 5-HT2C, 5-HT6 e 5-HT7) e α1-adrenérgicos. O aripiprazol é um representante único que atua como agonista parcial dos receptores D2 e antagonista 5-HT2, sendo desprovido de efeitos extrapiramidais, ganho de peso e hiperprolactinemia. Devido ao seu custo elevado em serviços de saúde pública, esse medicamento é geralmente introduzido na terapêutica apenas quando há intolerância aos efeitos colaterais dos demais fármacos. Os efeitos adversos mais comuns entre os antipsicóticos atípicos incluem ganho de peso, hiperlipidemia, hiperglicemia e indução de diabetes mellitus tipo 2, além de hiperprolactinemia moderada.
Estabilizadores de Humor
A mania, doença maníaco-depressiva ou transtorno bipolar, corresponde a uma desordem comportamental que causa mudanças incomuns no humor, energia, níveis de atividade e na capacidade de realizar tarefas diárias. Há uma carência de fármacos específicos para a terapêutica, sendo empregados agentes antipsicóticos, alguns anticonvulsivantes (ácido valproico, carbamazepina e lamotrigina) e o lítio (Li+).
As principais interações medicamentosas com o uso de Li+ estão ligadas a alterações na cinética de eliminação, já que há competição com o Na+ para a reabsorção e indução de efeitos tóxicos pela elevação dos níveis de Li+. Assim, diuréticos tiazídicos, como hidroclorotiazida, e, em menor extensão, espironolactona, amilorida e furosemida, reduzem a depuração do Li+. A toxicidade do Li+ é aumentada com haloperidol, inibidores seletivos da recaptação de serotonina e anticonvulsivantes, assim como a incidência de sintomas extrapiramidais com antipsicóticos.
Fármacos Ansiolíticos, Sedativos e Hipnóticos
A ansiedade é um estado emocional resultado de experiências vivenciadas, mas torna-se patológica quando desproporcional ou quando não há um objeto específico ao qual se direcione. A sedação refere-se a uma redução do nível de atividade do paciente, variando de um estado mínimo (o paciente responde a comando verbal e estimulação tátil leve) a um profundo (sem resposta a comando verbal). Os principais fármacos utilizados com esses propósitos são os benzodiazepínicos, barbitúricos e compostos “Z”, todos atuando em nível do receptor GABAA.
O ácido γ-aminobutírico (GABA) é o principal neurotransmissor inibitório do SNC, atuando em três tipos de receptores, sendo o GABAA o mais abundante. A ativação desse receptor pelo GABA promove a abertura do poro de um canal para Cl-, induzindo um influxo desse íon para os neurônios com consequente redução da excitabilidade por hiperpolarização da membrana neuronal. O receptor GABAA é formado por três subunidades diferentes (α, β e γ), sendo que a presença de todas essas subunidades e o subtipo da subunidade α determinam o perfil de ação dos benzodiazepínicos, barbitúricos e compostos “Z”.
Benzodiazepínicos
Todos os benzodiazepínicos atuam como moduladores alostéricos do GABA, aumentando a afinidade desse neurotransmissor pelo receptor GABAA e, consequentemente, intensificando o influxo de Cl- para os neurônios. Uma característica importante desses fármacos é que, mesmo em doses muito altas, não conseguem ativar os receptores GABAA na ausência de GABA, nem induzir anestesia cirúrgica quando administrados isoladamente. Quando utilizados como hipnóticos, os benzodiazepínicos reduzem a latência do sono, reduzem o tempo e aumentam o ciclo do sono com movimentos oculares rápidos (REM), prolongando o tempo total de sono.
Os principais efeitos colaterais dos benzodiazepínicos são fraqueza, visão borrada, vertigem, desconfortos intestinais, sonolência, prejuízo às habilidades psicomotoras, comprometimento cognitivo, hipotonia e aumento do risco de fratura. A maioria das interações medicamentosas clinicamente relevantes com fármacos benzodiazepínicos envolve a indução ou inibição de enzimas hepáticas ou efeitos aditivos com outros depressores do SNC, como o etanol.
Barbitúricos
Após a introdução dos benzodiazepínicos no mercado, houve uma larga substituição dos barbitúricos como fármacos sedativos e hipnóticos, tendo sua indicação principal atualmente como anticonvulsivantes e anestésicos. Os fármacos barbitúricos intensificam a ligação do GABA aos receptores GABAA, aumentando os períodos durante os quais esse canal permanece aberto. O resultado dessas características é a depressão em todos os graus do SNC. Como os barbitúricos, mesmo em pequenas doses, podem aumentar as reações aos estímulos dolorosos, deve-se evitar o uso desses fármacos em sedação com presença de dor. O efeito hipnótico desses fármacos é semelhante aos benzodiazepínicos, com diminuição da latência, aumento do tempo total de sono e redução do tempo do sono REM. No entanto, desenvolvem rápida tolerância e possuem um baixo índice terapêutico, podendo provocar intoxicação grave em casos de overdose, com risco de morte por depressão respiratória.
Compostos “Z”
Com a identificação de que a subunidade α do receptor GABAA pode conferir especificidade de ação pelos fármacos depressores do SNC, foi possível o desenvolvimento de fármacos hipnóticos seletivos. Os compostos “Z”, que compreendem zolpidem, zaleplona e zopiclona, atuam acentuando a corrente de Cl- pelo receptor GABAA, com maior seletividade aos que possuem a subunidade α1. Assim, esses fármacos atuam como hipnóticos de curta duração com reduzido aparecimento de efeitos residuais, sem atividade sedativa e ansiolítica significativa. O zolpidem é rapidamente absorvido após administração oral. Os efeitos adversos mais comuns desse fármaco incluem alterações gastrintestinais, dores de cabeça, desinibição e efeitos potenciais sobre a maioria das funções do SNC. A zaleplona tem afinidade similar ao zolpidem pelos receptores GABAA, mas possui duração de ação mais curta. É contraindicada em pacientes com insuficiência renal, hepática e respiratória, síndrome da apneia do sono e miastenia grave. Dor de cabeça é o efeito colateral mais relatado, com pouca ocorrência de náuseas, mialgia, dor abdominal e efeito residual.
Tratamentos dos Distúrbios Degenerativos do SNC
As doenças degenerativas do SNC são caracterizadas pela perda irreversível de neurônios localizados em determinadas regiões cerebrais, cuja fisiopatologia está intrinsecamente relacionada à neurotransmissão química. Dois exemplos proeminentes são a Doença de Parkinson e a Doença de Alzheimer.
Farmacoterapia da Doença de Parkinson
A Doença de Parkinson é um distúrbio crônico, progressivo, que afeta cerca de 1% das pessoas com mais de 60 anos de idade. Os achados patológicos mais proeminentes são a perda de neurônios dopaminérgicos na substância negra e a presença de inclusões intracelulares conhecidas como corpúsculos de Lewis. Os sintomas da doença surgem quando há uma redução da ativação dos receptores pós-sinápticos para dopamina D1 e D2. Além disso, como a acetilcolina antagoniza a neurotransmissão dopaminérgica no corpo estriado, o desbalanço desses dois neurotransmissores também contribui para o agravamento da doença. Diante disso, a primeira linha da farmacoterapia da Doença de Parkinson envolve a recuperação da atividade dopaminérgica, por exemplo, com o uso da levodopa ou agonistas dopaminérgicos, e a redução da neurotransmissão colinérgica.
A levodopa é o precursor metabólico imediato da dopamina, que consegue atravessar a barreira hematoencefálica utilizando um transportador de L-aminoácido e, ao alcançar os núcleos da base, é descarboxilada a dopamina. Os efeitos adversos mais frequentes incluem anorexia, náuseas, vômitos, arritmias cardíacas, discinesias e, em alguns pacientes, ataque de glaucoma agudo. As principais interações medicamentosas ocorrem com o uso de piridoxina (vitamina B6), que acelera o metabolismo extracerebral da levodopa, e com IMAO, que pode induzir crises hipertensivas.
Farmacoterapia da Doença de Alzheimer
A Doença de Alzheimer é caracterizada como uma desordem neurodegenerativa multifatorial que leva a uma progressiva perda da capacidade mental, comportamental, funcional e da capacidade de aprendizado. A neuropatologia dessa doença está relacionada ao acúmulo extracelular do peptídeo β-amiloide, formando oligâmeros solúveis que coalescem em fibrilas insolúveis, com consequente hiperfosforilação da proteína tau associada aos microtúbulos, disfunção neuronal e morte celular. Diante disso, a acentuação da neurotransmissão colinérgica remanescente, com a inibição das colinesterases nas fendas sinápticas e aumento da concentração da acetilcolina extracelular, corresponde ao tratamento de primeira linha.
Todos os anticolinesterásicos utilizados melhoram a cognição, comportamento e a funcionalidade em pacientes com Doença de Alzheimer leve a moderada. Os efeitos colaterais mais comuns são desconforto gastrintestinal, cãibras musculares e bradicardia. Em pacientes com Doença de Alzheimer moderada a grave, utilizam-se outros medicamentos como coadjuvantes terapêuticos, sendo a memantina o mais empregado. Esse fármaco é um antagonista não competitivo dos receptores para glutamato do tipo NMDA, atuando na manutenção da função normal glutamatérgica e reduzindo a taxa de deterioração clínica dos pacientes mais graves.
Fármacos Anticonvulsivantes
A epilepsia corresponde a uma enfermidade crônica caracterizada por episódios convulsivos recorrentes, gerados por uma descarga paroxística, hipersincrônica, excessiva e descontrolada de um grande número de neurônios. O tratamento farmacológico das epilepsias tem como base a inibição da despolarização neuronal anômala por meio da potencialização da ação do GABA ou da inibição da função dos canais para Na+ ou Ca2+.
Os fármacos anticonvulsivantes disponíveis podem causar uma variedade de efeitos indesejados, de tal forma que o aconselhado é tratar os pacientes com apenas um fármaco. Caso as convulsões não sejam controladas, a substituição por outro fármaco é preferível à adição de um novo. Alguns benzodiazepínicos, como clonazepam e diazepam, e o barbitúrico fenobarbital, também são utilizados no tratamento de todos os tipos de epilepsias.
A carbamazepina e a oxcarbazepina são utilizadas principalmente no tratamento das convulsões tônico-clônicas e crises parciais, com maior eficácia naquelas que iniciam no sistema límbico do que no córtex. Os efeitos indesejados do uso crônico da carbamazepina incluem sonolência, vertigem, ataxia, diplopia, visão borrada e redução da concentração plasmática de Na+. A fenitoína possui efeito anticonvulsivante sem causar depressão do SNC. Os efeitos adversos mais comuns ocorrem em pacientes idosos, como arritmias cardíacas e hipotensão. O ácido valproico é o anticonvulsivante mais utilizado por possuir amplo espectro de ação em todos os tipos de convulsões. A ação desse fármaco é atribuída à acentuada inativação dos canais para Na+ sensíveis à voltagem, redução das correntes de Ca2+ tipo T e alteração do metabolismo do GABA. As principais reações adversas envolvem desconfortos gastrintestinais e elevação das transaminases hepáticas.
Anestésicos Gerais
O estado de anestesia geral é definido como a perda da consciência e da reatividade a estímulos dolorosos intensos, induzida de maneira reversível pela ação de determinado fármaco em nível do SNC. Os anestésicos gerais causam depressão do SNC por acentuar as sinapses inibitórias e inibir as excitatórias, sendo que em concentrações elevadas podem induzir depressão profunda do centro respiratório no tronco cerebral.
O óxido nitroso, xenônio e cetamina atuam inibindo as respostas induzidas por receptores para glutamato do tipo NMDA, enquanto os demais anestésicos atuam potencializando a ação do GABA sobre os receptores GABAA. Outro local de ação envolve a ativação dos canais para K+ com domínio de dois poros, reduzindo a excitabilidade da membrana. Na indução anestésica, observa-se redução da pressão arterial sistêmica por ação vasodilatadora direta dos anestésicos, depressão do miocárdio e diminuição do tônus simpático central. A intubação endotraqueal é necessária para evitar aspiração devido à redução do esfíncter esofágico inferior e possibilidade de regurgitação. Geralmente, os anestésicos gerais agem sobre a zona de gatilho quimiorreceptora, induzindo náuseas e vômitos.
Anestésicos Intravenosos
Os anestésicos intravenosos induzem a perda da consciência em aproximadamente 20 segundos após chegarem ao cérebro, sendo preferidos para a indução da anestesia. Não são indicados para a manutenção da anestesia, com exceção do propofol quando empregado em infusão contínua. O tiopental é um barbitúrico altamente lipossolúvel com rápido início de ação. O tiopental possui pouco efeito analgésico e pode causar depressão respiratória e cardiovascular profunda. O propofol é o fármaco de escolha para indução da anestesia pelo fato de ser rapidamente metabolizado a conjugados inativos e possuir rápida taxa de distribuição. Contudo, o propofol pode induzir vasodilatação, bradicardia e depressão da respiração, podendo ser problemático em pacientes críticos. O etomidato é utilizado em procedimentos cirúrgicos rápidos, pois a recuperação da anestesia é mais rápida do que com o tiopental.
Anestésicos Inalatórios
Os anestésicos inalatórios são utilizados na prática clínica para a manutenção da anestesia induzida por um anestésico intravenoso. A principal via de administração e eliminação é a pulmonar. Assim, a potência de um anestésico inalatório é avaliada pela concentração alveolar mínima (CAM) capaz de inibir uma resposta motora a um estímulo doloroso. A profundidade da anestesia conseguida com um agente inalatório está relacionada com a pressão parcial que o anestésico alcança no cérebro, aproximando-se muito da pressão parcial no sangue arterial conseguida após a inalação.
Portanto, os principais fatores que afetam a pressão parcial do anestésico no sangue e cérebro são: (1) concentração do anestésico no ar inspirado; (2) ventilação pulmonar durante toda a anestesia; (3) o débito cardíaco e pulmonar e (4) solubilidade do anestésico no sangue. Todos os anestésicos reduzem a pressão arterial de maneira dependente da dose, principalmente o halotano e o enflurano que reduzem a contratilidade do miocárdio com risco de arritmias. A respiração também é deprimida de maneira dependente da dose até a apneia, principalmente pela inibição da resposta ventilatória à hipóxia e hipercapnia. Com exceção do óxido nitroso, os anestésicos inalatórios apresentam efeito relaxante muscular e potencializam o efeito dos bloqueadores neuromusculares.
Tabela Comparativa de Antidepressivos
| Classe de Fármaco | Mecanismo Principal | Principais Efeitos Adversos | Considerações Específicas |
|---|---|---|---|
| IMAO | Inibição da degradação de 5-HT e NA, aumentando sua concentração. | Hipotensão, tremores, insônia, ganho de peso, efeitos anticolinérgicos. Risco de síndrome serotonínica. | Interação grave com alimentos ricos em tiramina (crise hipertensiva) e outros fármacos serotoninérgicos. |
| ADT | Inibição da recaptação de 5-HT e NA, aumentando sua concentração. | Hipotensão ortostática, efeitos anticolinérgicos (boca seca, visão borrada, constipação, retenção urinária), sedação, cardiotoxicidade. | Ampla gama de usos além da depressão. Interação com etanol e anti-hipertensivos. |
| ISRS | Inibição seletiva da recaptação de 5-HT, aumentando sua concentração. | Distúrbios GI, ansiedade, disfunção sexual, prejuízo cognitivo. Menor incidência de efeitos anticolinérgicos e cardiovasculares. | Primeira escolha para depressão devido ao perfil de segurança. Risco de síndrome serotonínica, embora menor que IMAO. |
Perguntas Frequentes sobre Medicamentos do SNC
O que pode acontecer se eu parar abruptamente o tratamento com anticonvulsivantes?
A suspensão abrupta do tratamento com fármacos anticonvulsivantes pode ocasionar um aumento significativo na frequência e gravidade dos episódios de convulsão. Isso ocorre devido a um efeito de “rebote”, onde a neurotransmissão no cérebro se torna superestimulada, levando a convulsões mais descontroladas. É fundamental que qualquer alteração na medicação seja feita sob orientação médica rigorosa e, geralmente, de forma gradual.
É seguro para gestantes com epilepsia tomar anticonvulsivantes?
O tratamento de pacientes grávidas com epilepsia é de extrema cautela. Não é a epilepsia em si, mas o risco ao qual o feto está exposto devido a alguns anticonvulsivantes que preocupa. Por exemplo, podem ocorrer malformações como fenda palatina e lábio leporino, anomalias craniofaciais, defeitos cardíacos e do tubo neural (como espinha bífida). No entanto, é importante ressaltar que cerca de 90% das gestações de mulheres com epilepsia resultam em bebês saudáveis. A decisão de continuar ou ajustar a medicação durante a gravidez deve ser cuidadosamente avaliada por uma equipe médica especializada, pesando os riscos e benefícios para a mãe e o bebê.
Todos os anticonvulsivantes causam depressão do sistema nervoso central e exigem monitoramento rígido?
Não, nem todos os anticonvulsivantes apresentam efeitos depressores do SNC, e, portanto, o monitoramento em relação a esse aspecto não é sempre necessário. Embora alguns ansiolíticos, que porventura sejam usados na terapêutica do controle das convulsões, mereçam um controle mais rigoroso devido ao seu potencial depressor do SNC, muitos anticonvulsivantes atuam por mecanismos diferentes, como o bloqueio de canais para Na+ e Ca2+, ou a modulação de neurotransmissores como o glutamato e GABA, sem induzir depressão generalizada. A necessidade de monitoramento depende do fármaco específico e do perfil do paciente.
O tratamento com anticonvulsivantes é sempre para a vida toda?
Não, a descontinuidade do tratamento com anticonvulsivantes pode ser indicada em alguns casos, sempre a partir de uma avaliação médica criteriosa. Por exemplo, em situações onde o paciente permanece sem crises por um longo período e sob determinadas condições clínicas, o médico pode considerar a redução gradual e a eventual interrupção da medicação. No caso da amamentação, por exemplo, é aconselhada a interrupção de alguns fármacos anticonvulsivantes, dependendo do risco para o bebê e da necessidade materna. A decisão de descontinuar deve ser individualizada e jamais tomada sem acompanhamento profissional.
A compreensão da distribuição de medicamentos e da complexa farmacologia do sistema nervoso central é vital para o avanço da medicina e para a otimização dos tratamentos. A pesquisa contínua e a educação sobre esses temas são a chave para melhorar a saúde e a qualidade de vida dos pacientes.
Se você quiser conhecer outros artigos parecidos com O Movimento Essencial dos Medicamentos no Corpo, pode visitar a categoria Farmacologia.