09/05/2022
A eficácia de um medicamento não depende apenas de sua capacidade de combater uma doença, mas também de como ele interage com o nosso organismo. Essa interação complexa é estudada pela farmacologia, uma ciência que se divide em dois pilares fundamentais: a farmacodinâmica e a farmacocinética. Enquanto a primeira se aprofunda nos mecanismos de ação dos fármacos, revelando como eles produzem seus efeitos terapêuticos, a segunda desvenda o que o corpo faz com o medicamento, desde o momento em que ele entra no sistema até sua completa eliminação. Compreender esses processos é crucial para garantir que a dose certa atinja o local certo na concentração ideal, transformando uma promessa terapêutica em um resultado real para a saúde do paciente.
- Farmacocinética vs. Farmacodinâmica: Compreendendo a Essência da Ação Medicamentosa
- A Absorção de Fármacos: A Porta de Entrada no Organismo
- Distribuição dos Fármacos: O Caminho para o Alvo
- Metabolismo e Excreção: A Eliminação do Organismo
- Biodisponibilidade e Depuração (Clearance): Métricas Essenciais
- Perguntas Frequentes (FAQ)
- Conclusão
Farmacocinética vs. Farmacodinâmica: Compreendendo a Essência da Ação Medicamentosa
No universo da farmacologia, a distinção entre farmacocinética e farmacodinâmica é fundamental para entender a jornada de um medicamento no corpo. A farmacodinâmica é o estudo do mecanismo de ação dos fármacos, ou seja, como eles interagem com as células e tecidos para produzir um efeito biológico. É o que o medicamento faz ao corpo.
Por outro lado, a farmacocinética avalia os efeitos que o corpo faz com o fármaco. Ela abrange quatro processos essenciais, conhecidos pela sigla ADME: Absorção, Distribuição, Metabolismo e Excreção. Como bem destaca Alexandre Massao Sugawara, farmacêutico e professor do ICTQ, mesmo a mais promissora das drogas fracassará em seus intuitos terapêuticos se não alcançar concentrações mínimas efetivas no órgão alvo para exercer seus efeitos. A farmacocinética, portanto, é a chave para garantir que o fármaco chegue ao seu destino e permaneça lá em quantidade suficiente para agir.
Tabela Comparativa: Farmacocinética vs. Farmacodinâmica
| Aspecto | Farmacocinética | Farmacodinâmica |
|---|---|---|
| Foco Principal | O que o corpo faz com o fármaco | O que o fármaco faz ao corpo |
| Processos Chave | Absorção, Distribuição, Metabolismo, Excreção (ADME) | Mecanismo de ação, Efeitos terapêuticos, Efeitos adversos |
| Objetivo | Determinar a concentração do fármaco no local de ação ao longo do tempo | Descrever a relação entre a concentração do fármaco e a resposta clínica |
| Exemplo de Pergunta | Quão rápido o fármaco é absorvido? | Como o fármaco reduz a dor? |
A Absorção de Fármacos: A Porta de Entrada no Organismo
Para que um fármaco comece sua jornada terapêutica, ele precisa primeiro ser absorvido pelo corpo. Esse processo envolve a transposição de barreiras teciduais, que são compostas, em última análise, por membranas celulares de natureza lipoproteica. A capacidade de um composto químico se difundir através dessas membranas depende fundamentalmente de suas propriedades físico-químicas, especialmente sua miscibilidade em um meio predominantemente oleoso.
Características Físico-Químicas Cruciais
A lipossolubilidade de um fármaco é um dos fatores mais determinantes para sua absorção. O coeficiente de partição octanol/água (P) é um indicador crucial dessa tendência, e seu logaritmo, o LogP, quantifica essa propriedade. Um LogP > 0 indica que a molécula é lipofílica (afinidade por gordura), enquanto um LogP < 0 indica que é hidrofílica (afinidade por água). Valores muito baixos de LogP dificultam a permeação pelas membranas celulares, e valores muito elevados podem reter as moléculas na membrana devido à sua alta lipossolubilidade. O valor ideal de LogP para a maioria dos fármacos situa-se entre 2 e 5. Curiosamente, uma molécula precisa de certa lipossolubilidade para atravessar as barreiras e, ao mesmo tempo, de certa hidrossolubilidade para se distribuir pelos líquidos corporais, já que nosso corpo é cerca de 70% água. A química farmacêutica frequentemente busca criar compostos ionizáveis que possam ter ambas as propriedades em diferentes faixas de pH.
O Papel do pH e da Ionização
A ionização de um fármaco, influenciada pelo pH do ambiente, é um fator crítico para sua absorção. Fármacos ácidos fracos, como a aspirina, em um ambiente altamente ácido (como o suco gástrico), predominam em sua forma protonada, não ionizada e lipossolúvel, o que lhes permite atravessar as membranas biológicas da parede do estômago com mais facilidade. Ao atingir o plasma, que é mais alcalino, a aspirina é desprotonada, tornando-se ionizada e aumentando sua hidrossolubilidade, facilitando sua dissolução no plasma e evitando sua reabsorção retrógrada.
A Equação de Henderson-Hasselbalch descreve essa relação matemática entre o pKa de um fármaco (ácido ou básico) e o pH do meio biológico. Por exemplo, um fármaco ácido fraco com pKa 6,0 no estômago (pH 1,0) estará predominantemente em sua forma não ionizada (lipossolúvel), favorecendo a absorção. Ao chegar ao plasma (pH 7,4, ou para simplificar, 7,0), a situação se inverte, e a forma ionizada (hidrossolúvel) passa a predominar, permitindo sua distribuição no sangue.
Barreiras Fisiológicas e Transportadores Proteicos
Nem todos os fármacos se difundem através das membranas por simples difusão. Alguns utilizam transportadores proteicos, como é o caso de fármacos peptídicos e aminoácidos, que se valem dos mesmos transportadores de seus análogos nutricionais. Um exemplo notável é a levodopa, utilizada no tratamento do Parkinson. Atualmente, superfamílias de transportadores de membrana, como a ATP binding cassete (ABC), Solute Carrier (SLC) e Organic Solute Carrier (SLCO), são extensivamente estudadas. Elas são encontradas em diversas barreiras biológicas, incluindo intestino, fígado, barreira hematoencefálica, placenta, glândulas mamárias e rins. O DrugBank© (https://www.drugbank.ca) é uma excelente fonte de consulta para informações sobre esses transportadores e suas interações.
A glicoproteína-P (P-gP) é um transportador de efluxo de grande importância, presente em inúmeras membranas biológicas, como no intestino e na barreira hematoencefálica. Ela atua como um mecanismo de proteção, eliminando xenobióticos do ambiente intracelular. A cafeína, por exemplo, é um inibidor da P-gP, e seu uso concomitante com fármacos substratos da P-gP pode aumentar a concentração desses compostos no sangue. Variações genéticas na expressão da P-gP podem explicar a não absorção de certos fármacos, como a digoxina.
Vias de Administração: Escolhas Estratégicas
A via pela qual um medicamento é administrado é um fator crucial que influencia diretamente sua absorção. A insulina, por ser uma molécula proteica, seria digerida se administrada por via oral, por isso, sua via de administração é injetável (parenteral). A via parenteral é vantajosa por permitir que o fármaco alcance rapidamente picos plasmáticos, o que é essencial em emergências. No entanto, a via oral se destaca pela conveniência, facilidade de adesão do paciente e baixo custo operacional. Outras vias, como a transdérmica e a inalatória, são indicadas para doenças específicas da pele e do sistema respiratório, respectivamente, devido à baixa absorção sistêmica, minimizando efeitos colaterais em outras partes do corpo.
Onde Ocorre a Absorção dos Medicamentos? Uma Jornada Pelo Trato Gastrointestinal
Para um fármaco administrado por via oral, a absorção é um desafio. Ele deve sobreviver a ambientes com pH baixo e a secreções gastrointestinais (GI) que podem degradá-lo. Fármacos peptídicos, como a insulina, são particularmente vulneráveis. A absorção oral envolve o transporte através das membranas das células do trato GI e é influenciada por diversos fatores:
- Diferenças no pH do lúmen ao longo do trato GI.
- Área de superfície disponível para absorção.
- Perfusão sanguínea no local.
- Presença de bile e muco.
- Natureza das membranas epiteliais.
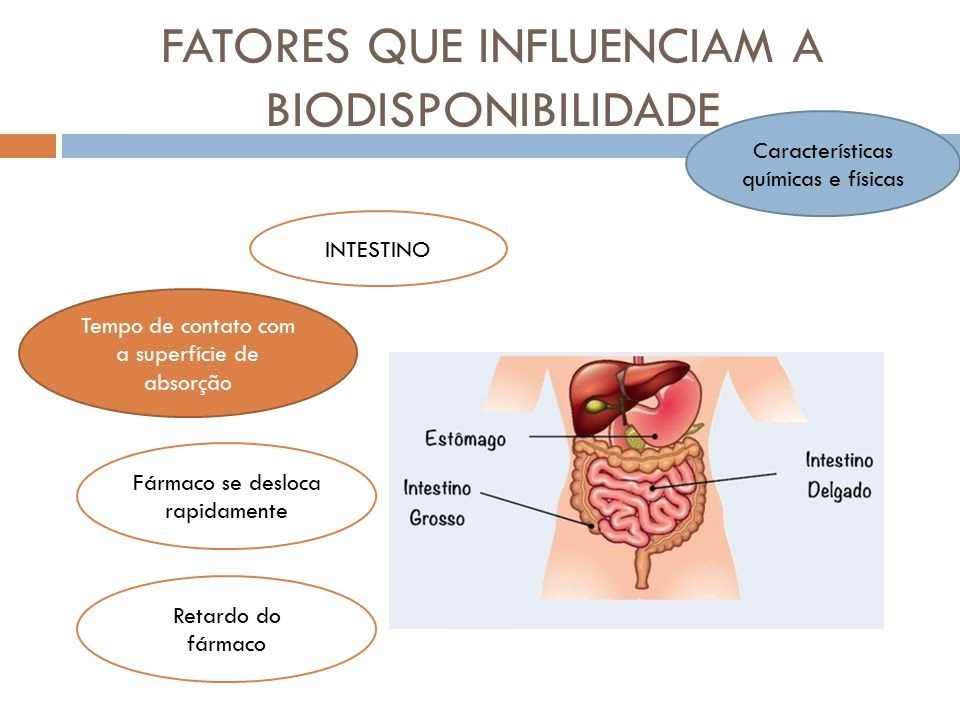
A mucosa oral, com seu epitélio fino e rica vascularização, favorece a absorção, mas o contato é geralmente muito breve. Fármacos colocados entre a gengiva e a bochecha (bucal) ou sob a língua (sublingual) são retidos por mais tempo, potencializando a absorção.
O estômago é o primeiro órgão de contato intenso para fármacos orais. Embora tenha uma superfície epitelial relativamente grande, sua espessa camada de muco e o curto tempo de trânsito limitam a absorção. O esvaziamento gástrico é, muitas vezes, o passo limitante da velocidade para a absorção da maioria dos fármacos, especialmente porque a maior parte da absorção ocorre no intestino delgado. Alimentos, em particular os gordurosos, diminuem o esvaziamento gástrico, o que explica por que alguns fármacos são absorvidos mais rapidamente com o estômago vazio. Fármacos que afetam o esvaziamento gástrico também podem alterar a taxa de absorção de outros medicamentos.
O intestino delgado possui a maior área de superfície para absorção de fármacos no trato GI, e suas membranas são mais permeáveis que as do estômago. Por isso, a maioria dos fármacos é absorvida principalmente aqui. O pH no lúmen varia de 4 a 5 no duodeno e se torna mais alcalino (próximo de 8) no íleo distal. A flora gastrointestinal e a diminuição do fluxo sanguíneo (como no choque) podem reduzir a absorção. O tempo de trânsito intestinal também é crucial, especialmente para fármacos absorvidos por transporte ativo, aqueles que se dissolvem lentamente ou os que são mais polares.
A Influência da Forma Farmacêutica
A forma farmacêutica do medicamento, como comprimidos ou cápsulas, é um fator determinante. Para que as formas sólidas sejam absorvidas, elas precisam primeiro se dissolver. A taxa de dissolução pode ser o passo limitante da velocidade de absorção. A manipulação da formulação, por exemplo, a forma do fármaco como sal, cristal ou hidrato, pode alterar essa taxa e, consequentemente, controlar a absorção total.
Distribuição dos Fármacos: O Caminho para o Alvo
Após a absorção, o fármaco se distribui pelo organismo. O grau de distribuição nos tecidos depende da sua ligação às proteínas plasmáticas e aos próprios tecidos. Na corrente sanguínea, os fármacos existem em duas formas: livre (sem ligação) e ligada reversivelmente a componentes sanguíneos, como proteínas plasmáticas e células sanguíneas. As proteínas plasmáticas mais importantes para a ligação de fármacos são a albumina, a alfa-1-glicoproteína ácida e as lipoproteínas. Fármacos ácidos tendem a se ligar mais à albumina, enquanto fármacos básicos se ligam mais à alfa-1-glicoproteína ácida ou às lipoproteínas.
É crucial notar que apenas o fármaco livre está disponível para difusão passiva para os locais extravasculares ou teciduais onde ocorrem os efeitos farmacológicos. Assim, a concentração do fármaco livre na circulação sistêmica é o que classicamente determina a concentração no local ativo e, consequentemente, sua eficácia. Em concentrações elevadas do fármaco, os sítios de ligação podem saturar, o que pode levar a interações de deslocamento entre fármacos, aumentando a fração livre de um deles e, potencialmente, seus efeitos tóxicos, embora isso seja menos comum na prática clínica.
Os fármacos também podem se acumular em tecidos ou compartimentos corporais, o que pode prolongar sua ação, pois os tecidos liberam o fármaco à medida que sua concentração plasmática diminui. Um exemplo clássico é o tiopental, um anestésico altamente solúvel em lipídios. Após uma injeção intravenosa, ele penetra rapidamente no encéfalo, produzindo um efeito anestésico intenso e rápido. No entanto, esse efeito é breve, pois o fármaco é rapidamente redistribuído para tecidos adiposos com menor perfusão. O tiopental é então liberado lentamente do depósito de gordura, mantendo níveis plasmáticos subanestésicos. Se doses repetidas forem administradas, grandes quantidades podem ser armazenadas no tecido adiposo, prolongando o efeito. Outro exemplo é a cloroquina, que pode se acumular em leucócitos e hepatócitos em concentrações mil vezes maiores que no plasma.
Metabolismo e Excreção: A Eliminação do Organismo
Após a distribuição, os fármacos são submetidos a processos de metabolização e, posteriormente, eliminação, que levam ao declínio da sua concentração plasmática.
Biotransformação (Metabolismo)
As reações de metabolização são classificadas em dois tipos principais:
- Reações de Oxidação/Redução: Modificam a estrutura química do fármaco. O sistema do citocromo P450, localizado principalmente no fígado, é um conjunto de famílias de enzimas que realizam essas reações.
- Reações de Conjugação/Hidrólise: Hidrolisam o fármaco ou o conjugam com uma molécula grande e polar. O objetivo é geralmente inativar a substância ou, mais comumente, aumentar sua solubilidade para facilitar a excreção na urina ou na bile. Em alguns casos, a hidrólise ou conjugação pode ativar pró-fármacos. Grupos comumente adicionados incluem glicuronato, sulfato, glutationa e acetato.
Os efeitos dessas reações podem ser alterados pela presença de outros fármacos. Barbitúricos, por exemplo, são poderosos indutores de enzimas do citocromo P450, enquanto outros fármacos podem inibi-las. A compreensão dessas interações medicamentosas é essencial para a dosagem apropriada e segura. Sugawara recomenda o DrugBank© e a Micromedex© como fontes de informação sobre substratos, inibidores e indutores das enzimas do citocromo P450.
Excreção Renal: O Descarte do Fármaco
A excreção do fármaco inicia-se após a biotransformação ou em paralelo a ela, e pode ser modificada por fatores que alteram a filtração glomerular, a reabsorção e a secreção tubular. A taxa de filtração glomerular (TFG) influencia diretamente a depuração do fármaco. Fármacos que se ligam fortemente a proteínas plasmáticas têm suas taxas de filtração diminuídas, pois as proteínas não são filtráveis pelo glomérulo.
Após a filtração, o processamento tubular do filtrado envolve dois processos: reabsorção e secreção tubular. A reabsorção de fármacos nos túbulos depende do pH intraluminal. Um pH ácido no filtrado favorece a reabsorção de fármacos ácidos de volta ao corpo. Por exemplo, a alcalinização da urina é uma estratégia para "sequestrar" a aspirina (um ácido fraco) nos túbulos, aumentando sua excreção em casos de overdose.
Alguns fármacos são secretados diretamente para dentro dos túbulos renais por mecanismos de transporte de ânions e cátions orgânicos. Os transportadores de ânions orgânicos, por exemplo, permitem a secreção de muitos antibióticos (cefalosporinas, penicilinas), diuréticos (furosemida) e analgésicos. A probenecida e o lítio podem inibir esse sistema, diminuindo a excreção de fármacos que o utilizam. O sistema de transporte catiônico é responsável pela secreção de bases fracas, como cimetidina, metadona e morfina. A P-glicoproteína (P-gP) também atua como uma bomba de efluxo que secreta múltiplos cátions orgânicos, como a digoxina, e seu transporte pode ser inibido por quinidina, verapamil e ciclosporina A, resultando em importantes interações medicamentosas.
Biodisponibilidade e Depuração (Clearance): Métricas Essenciais
A biodisponibilidade é um conceito crucial que indica a fração do fármaco que é absorvida e chega à circulação sistêmica de forma inalterada. Ela é o resultado da via de administração, das propriedades físico-químicas do fármaco e de fatores individuais do paciente, como a presença de transportadores. É apresentada sob a fórmula: Biodisponibilidade = (AUC oral / AUC IV) × 100, onde AUC é a Área Sob a Curva de concentração plasmática versus tempo.
A depuração (clearance) de um fármaco é um parâmetro que integra todos os fenômenos farmacocinéticos de metabolização e excreção. Ela indica a capacidade do organismo de eliminar o fármaco e é um indicador mais significativo do tempo de ação do fármaco em seus alvos moleculares, celulares e orgânicos. Ao diminuir a concentração do fármaco ativo no sangue, o metabolismo e a excreção reduzem o tempo durante o qual um fármaco é capaz de atuar sobre um órgão alvo.
A meia-vida de eliminação é definida como o tempo necessário para que a concentração do fármaco no plasma diminua pela metade. O conhecimento da meia-vida permite calcular a frequência de doses necessária para manter a concentração plasmática dentro da faixa terapêutica. Fatores como a diminuição da massa muscular (comum no envelhecimento) podem minimizar o volume de distribuição e, consequentemente, a meia-vida do fármaco, enquanto a obesidade pode aumentá-la. Tipicamente, os esquemas posológicos ótimos buscam manter a concentração plasmática do fármaco em um estado de equilíbrio dinâmico dentro de sua janela terapêutica. Esse estado é alcançado quando a taxa de aporte do fármaco é igual à sua eliminação, o que geralmente ocorre após cerca de quatro a cinco meias-vidas.
Perguntas Frequentes (FAQ)
Qual é a diferença entre farmacocinética e farmacodinâmica?
A farmacocinética estuda o que o corpo faz com o fármaco (Absorção, Distribuição, Metabolismo, Excreção), enquanto a farmacodinâmica estuda o que o fármaco faz ao corpo (seu mecanismo de ação e efeitos).
Onde são absorvidos os medicamentos?
Os medicamentos podem ser absorvidos em diversos locais, dependendo da via de administração. Para a via oral, a absorção ocorre principalmente no intestino delgado, devido à sua grande área de superfície e membranas mais permeáveis. No entanto, uma pequena absorção pode ocorrer na mucosa oral e no estômago, embora este último seja limitado por sua espessa camada de muco e curto tempo de trânsito. Outras vias de administração, como a injetável, transdérmica ou inalatória, têm seus próprios locais de absorção específicos (músculos, pele, pulmões, respectivamente).
Como é que os fármacos são transportados no sangue?
No sangue, os fármacos são transportados de duas formas: como fármaco livre (não ligado) em solução ou ligados reversivelmente a componentes sanguíneos, principalmente proteínas plasmáticas como a albumina, a alfa-1-glicoproteína ácida e as lipoproteínas. Apenas a fração livre do fármaco está disponível para exercer seus efeitos farmacológicos nos tecidos.
Quais são os fatores que influenciam na absorção dos medicamentos?
Diversos fatores influenciam a absorção dos medicamentos, incluindo: características físico-químicas da droga (como lipossolubilidade e grau de ionização), o veículo utilizado na formulação, a perfusão sanguínea no local de absorção, a área de superfície de absorção à qual o fármaco é exposto, a via de administração escolhida e a forma farmacêutica do medicamento.
Conclusão
A jornada de um medicamento no corpo humano é um processo intrincado e fascinante, regido pelos princípios da farmacocinética e da farmacodinâmica. Desde o momento da absorção até a sua completa eliminação, cada etapa é influenciada por uma miríade de fatores, desde as propriedades moleculares do fármaco até as características individuais do paciente. A compreensão aprofundada desses processos não é meramente acadêmica; ela é a base para a prática clínica segura e eficaz.
É por tudo isso que o conhecimento prático em farmacocinética clínica e farmacodinâmica se torna indispensável para o farmacêutico moderno. Como enfatiza o professor Sugawara, este profissional possui uma capacidade ímpar de intervir e otimizar a terapia medicamentosa, garantindo a plena efetividade e segurança dos tratamentos. Não é mais concebível que um profissional de saúde tão capacitado permaneça subutilizado. O farmacêutico deve, de fato, sair de trás do balcão, levantar-se da cadeira de escritório e ir ao encontro das necessidades da população, demonstrando a amplitude de suas competências e exercendo plenamente seu papel social na promoção da saúde.
Se você quiser conhecer outros artigos parecidos com Farmacocinética: A Jornada do Medicamento, pode visitar a categoria Farmacologia.